. B. Motor neuron filiform cytoplasmic inclusion is revealed by anti-TDP-43 in the spinal cord (black arrow). Other remaining motor neurons show loss of normal nuclear staining (red arrows). C. Many cortical neuronal small para nuclear inclusions are detected with antibodies to TDP-43 (black arrows). Short cortical dystrophic neurites are observed with anti-TDP-43 (black arrowheads) Loss of normal nuclear staining of TDP-43 is noted (red arrows). D. Some hippocampal neuronal cytoplasmic inclusions in dentate granule cells are displayed (black arrows). Loss of normal nuclear staining of TDP-43 is noted (red arrow). Magnification x400. Journal Pre-proof")
A. Marked depletion of motor neurons from the anterior horn of the spinal cord (H&E). B. Motor neuron filiform cytoplasmic inclusion is revealed by anti-TDP-43 in the spinal cord (black arrow). Other remaining motor neurons show loss of normal nuclear staining (red arrows). C. Many cortical neuronal small para nuclear inclusions are detected with antibodies to TDP-43 (black arrows). Short cortical dystrophic neurites are observed with anti-TDP-43 (black arrowheads) Loss of normal nuclear staining of TDP-43 is noted (red arrows). D. Some hippocampal neuronal cytoplasmic inclusions in dentate granule cells are displayed (black arrows). Loss of normal nuclear staining of TDP-43 is noted (red arrow). Magnification x400.
Introduction
The variant c.1414-1G > T in theGRN gene has been previously reported as probably pathogenic in subjects of Hispanic origin in the American continent.
Methods
We report five families of Spanish origin carrying this variant, including the clinical, neuroimaging, and laboratory findings.
Results
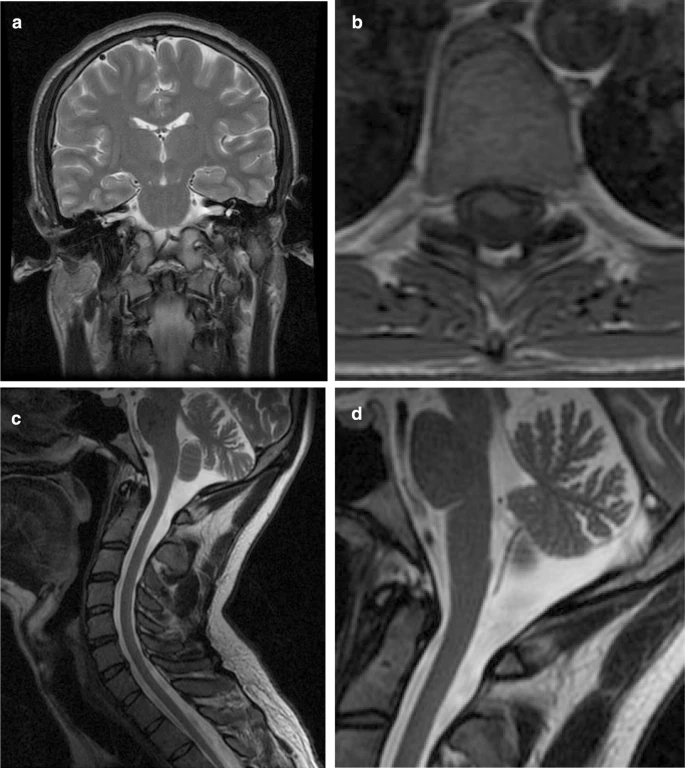
Phenotypes were strikingly different, including cases presenting with behavioral variant of frontotemporal dementia, with semantic variant of primary progressive aphasia, with rapidly progressive motor neuron disease (pathologically documented), and with tremor-dominant parkinsonism. Retinal degeneration has been found in homozygous carriers only. Ex vivo splicing assays confirmed that the mutation c.1414-1G > T affects the splicing of the exon, causing a loss of 20 amino acids in exon 11.
Conclusions
We conclude that variant c.1414-1G > Tn the GRN gene is pathogenic, can lead to a variety of clinical presentations and to gene dosage effect, and probably has a Spanish founder effect.
Full text: https://doi.org/10.1016/j.nrleng.2022.10.001