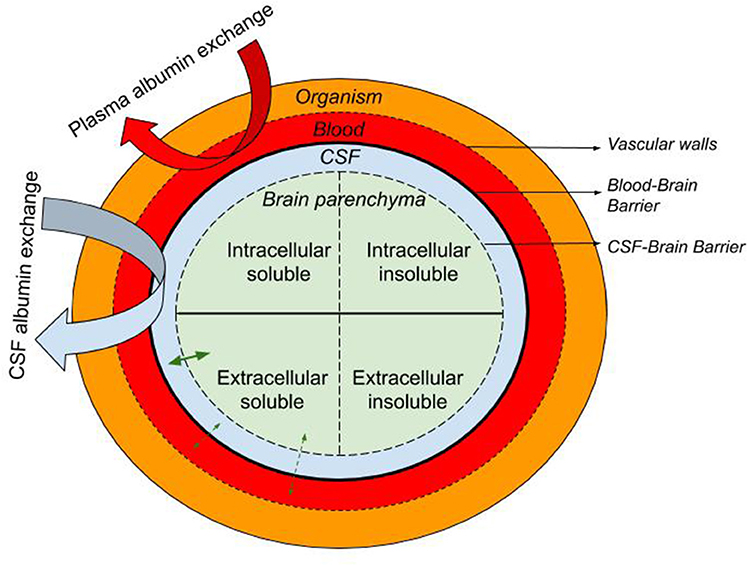
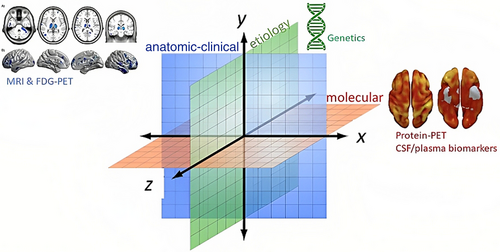
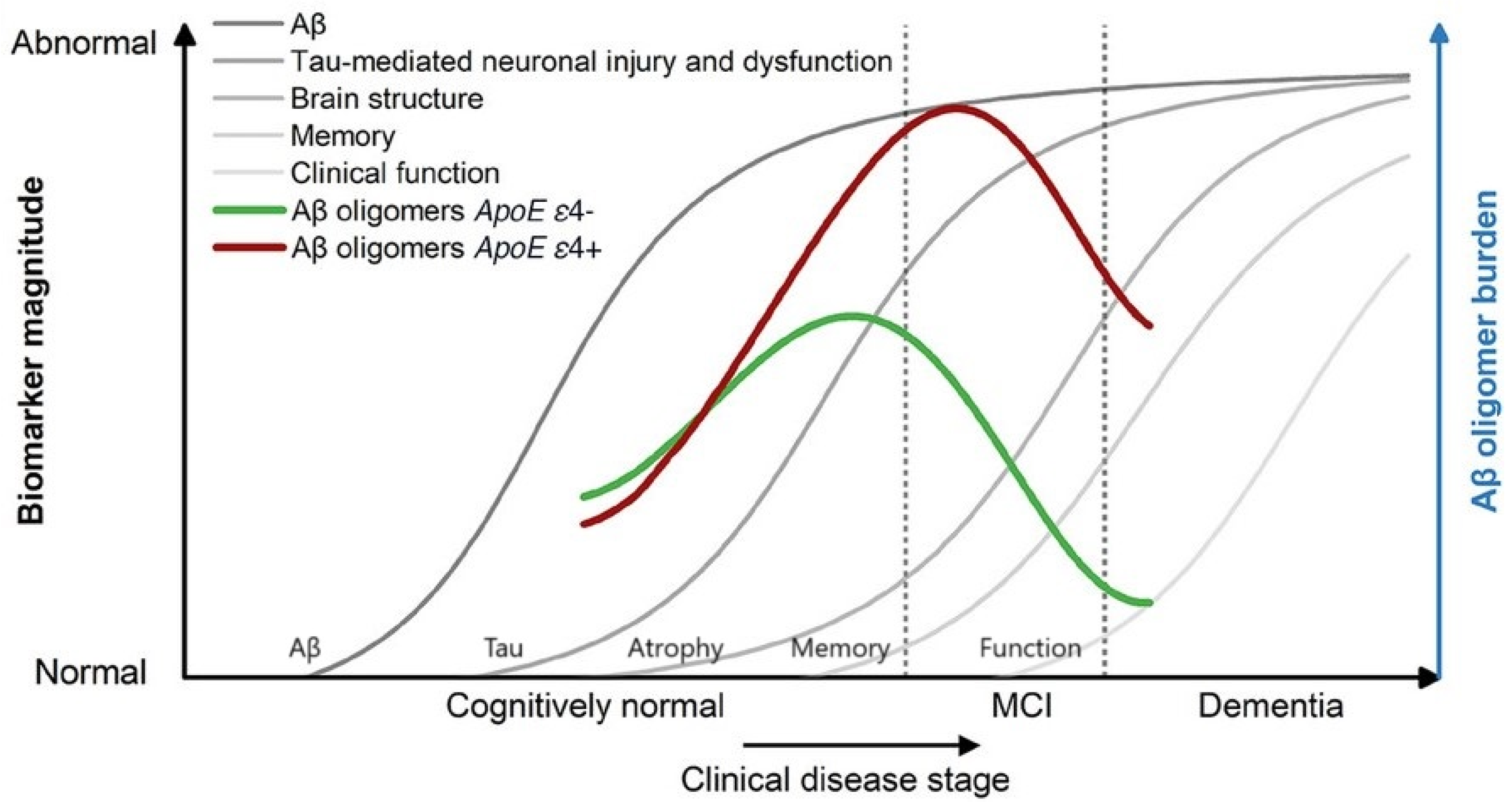
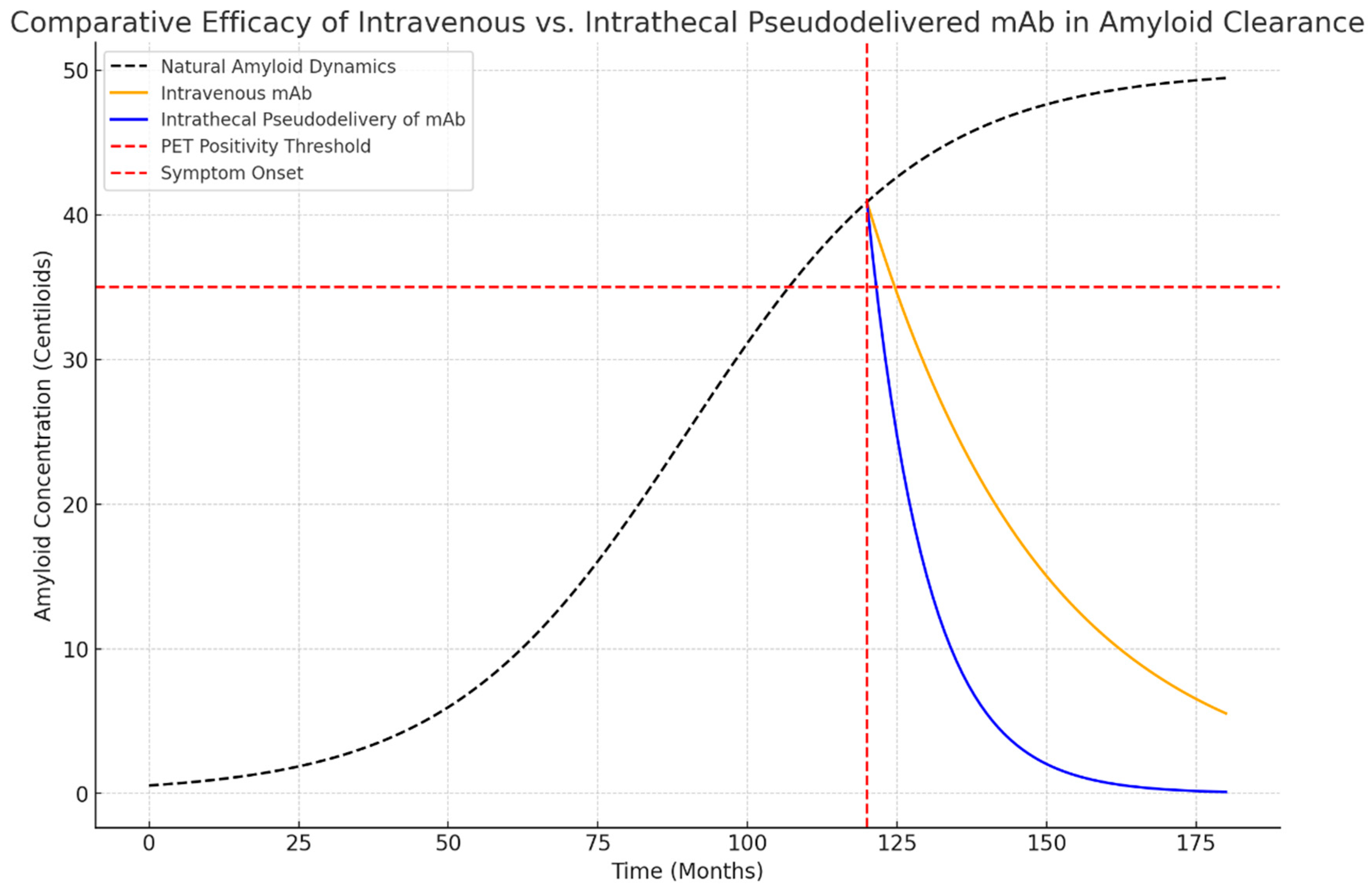
I am pleased to announce that I am editing a new volume titled "Intrathecal Drug Delivery", which will be published as part of the NEUROMETHODS series by Springer Nature. This book aims to provide a comprehensive and cutting-edge overview of intrathecal drug delivery, covering clinical applications, technological advancements, and experimental approaches.
We are currently assembling a diverse group of international experts in neurology, neurosurgery, anesthesiology, biomedical engineering, and related fields to contribute chapters to this volume. If you or a colleague are working on intrathecal drug delivery and would like to contribute, we invite you to express your interest!
Topics Already Confirmed in the Book Include:
✅ Existing and upcoming intrathecal drugs
✅ Parameters influencing drug distribution in the CSF
✅ Continuous and controlled intrathecal drug delivery
✅ Intrathecal drug delivery for chronic pain
✅ Investigating intrathecal treatment of leptomeningeal disease
✅ Pumpless implantable devices for intrathecal pseudodelivery of drugs
... and more!
Who Should Contribute?
We are looking for researchers, clinicians, and industry professionals with expertise in:
🔹 Neurology and Neurosurgery
🔹 Pain Medicine and Anesthesiology
🔹 Biomedical Engineering and Drug Delivery Systems
🔹 Neuropharmacology and Experimental Models of Intrathecal Therapies
Key Information for Authors:
📌 Manuscript submission deadline: May 31, 2025
📌 Chapter length: ~30–60 pages
📌 No cost for authors – Springer covers all publishing expenses
📌 Figures (including color images) are free of charge
📌 Global visibility – Springer Nature ensures high discoverability
If you are interested in contributing or know someone who might be a good fit, please reach out to me! This is a great opportunity to showcase your work in an internationally recognized book series.
Let’s shape the future of intrathecal drug delivery research together! 🚀
For inquiries or to propose a chapter, contact me at: menendezgmanuel @ uniovi.es
📢 Please share this post with colleagues who might be interested!
#IntrathecalDrugDelivery #Neuropharmacology #Neurosurgery #Neurology #PainMedicine #BiomedicalEngineering #DrugDelivery #SpringerNature #NEUROMETHODS #MedicalResearch


. B. Motor neuron filiform cytoplasmic inclusion is revealed by anti-TDP-43 in the spinal cord (black arrow). Other remaining motor neurons show loss of normal nuclear staining (red arrows). C. Many cortical neuronal small para nuclear inclusions are detected with antibodies to TDP-43 (black arrows). Short cortical dystrophic neurites are observed with anti-TDP-43 (black arrowheads) Loss of normal nuclear staining of TDP-43 is noted (red arrows). D. Some hippocampal neuronal cytoplasmic inclusions in dentate granule cells are displayed (black arrows). Loss of normal nuclear staining of TDP-43 is noted (red arrow). Magnification x400. Journal Pre-proof")